Since v.5.0, there are two ways of calculating electron-phonon coefficients, distinguished according to the value of variable electron_phonon. The following holds for the case electron_phonon= 'interpolated' (see also Example 03).
The calculation of electron-phonon coefficients in metals is made difficult by the slow convergence of the sum at the Fermi energy. It is convenient to use a coarse k-point grid to calculate phonons on a suitable wavevector grid; a dense k-point grid to calculate the sum at the Fermi energy. The calculation proceeds in this way:
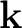 -point grid (or a scf calculation
followed by a non-scf one on the dense -point grid); specify
option la2f=.true. to pw.x in order to save a file with
the eigenvalues on the dense k-point grid. The latter MUST contain
all and
+
-point grid (or a scf calculation
followed by a non-scf one on the dense -point grid); specify
option la2f=.true. to pw.x in order to save a file with
the eigenvalues on the dense k-point grid. The latter MUST contain
all and
+ 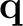 grid points used in the subsequent
electron-phonon
calculation. All grids MUST be unshifted, i.e. include = 0.
grid points used in the subsequent
electron-phonon
calculation. All grids MUST be unshifted, i.e. include = 0.
See the appendix for the relevant formulae. Important notice: the q→ 0 limit of the contribution to the electron-phonon coefficient diverges for optical modes! please be very careful, consult the relevant literature.